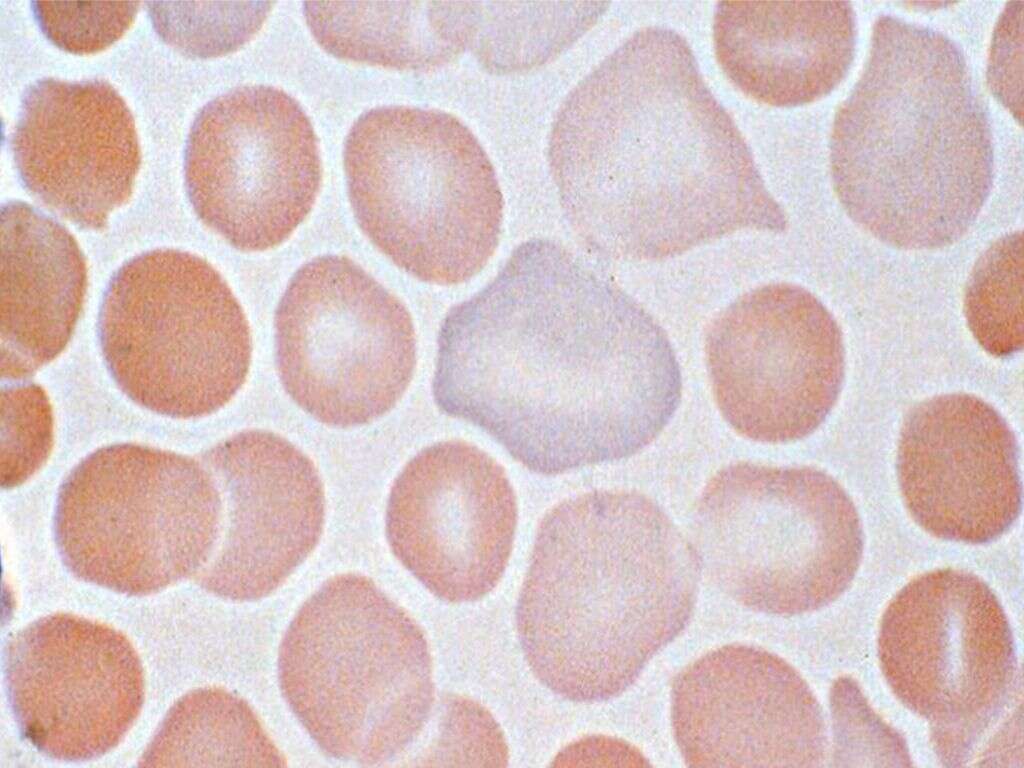
10 Sickle Cell Anemia Symptoms


Sickle cell anemia is a genetic disorder that results in the mutation of a component (β chain) of hemoglobin, an oxygen-carrying protein that can be found in red blood cells (RBCs). It is an autosomal recessive condition which means it occurs when the individual inherits one abnormal copy of the hemoglobin gene from each parent (resulting in a total of two copies of abnormal genes). This gene can be found on chromosome 11. In “stressful” conditions for the RBCs, the mutated hemoglobin (HbS) polymerizes and aggregates resulting in sickle-shaped RBCs (i.e. the shape of a reaping hook). This causes the red blood cell to continuously sickle and de-sickle, resulting in damage to its outer membrane. Thus, organs like the spleen (part of the reticuloendothelial system) remove these damaged RBCs from the circulation. However, RBCs can also dehydrate and rupture inside blood vessels (intravascular hemolysis) or produce occlusion of blood vessels and subsequent infarction of tissues (obstruction of blood supply and death of the tissue).
Symptoms of sickle cell anemia usually start around 5 to 6 months of age. An episode of sickle cell attack known as sickle cell crisis can be triggered by stress, dehydration, temperature changes, and high altitude. An individual with only one abnormal copy of the gene does not usually have symptoms and is said to have the sickle cell trait, otherwise known as carriers. The diagnosis can be made by screening for the mutated hemoglobin (HbS). Screening for this disease at birth is mandatory in many countries.
Management of the disorder includes the prevention of infection through vaccination, the use of pain medication for crisis, high fluid intake, and antibiotics. As of 2015, it has been estimated that 4.4 million individuals have sickle cell disease with an additional 43 million carriers. Approximately 80 percent of cases occur in sub-Saharan Africa. It is also frequently found in regions of the Arabian Peninsula, parts of India, and in people of African ethnicity in other parts of the world. This is likely due to the protective role of the disease against malaria caused by plasmodium falciparum.
Symptom #1: Pain
In sickle cell crises, periodic episodes of pain can occur. The pain occurs when the sickle-shaped red blood cells cause a blockage in the flow of blood through the tiny blood vessels to the joints, chest, abdomen, and more. Pain can also be experienced in the extremities, related to long bones. Varying in intensity, pain that occurs during sickle cell crisis can last from a few hours to a several days.
While some patients experience only a few pain episodes per year, others can suffer a lot more. Severe cases of sickle cell pain crisis may require hospital admission.
Symptom #2: Fatigue
Fatigue is a sensation of tiredness that encompasses a range of complaints including malaise, lethargy, lassitude, and exhaustion. It is a nonspecific symptom; thus it can be observed in many conditions.
In sickle cell disease, the RBCs rupture easily either in the interior of blood vessels (intravascular hemolysis) or in organs of the reticuloendothelial system, such as the spleen (extravascular hemolysis). This results in anemia, where there is a decrease in the mass of red blood cells that can carry the oxygen supply for all the tissues in the body. A common symptom of anemia is fatigue.
Symptom #3: Dactylitis
Dactylitis refers to the inflammation of a finger or toe (an entire digit). It can be a painful condition. Dactylitis can be seen in conditions such as ankylosing spondylitis, psoriatic arthritis, tuberculosis, leprosy, syphilis, sarcoidosis, and sickle cell disease. In patients with sickle cell disease, dactylitis occurs as a result of a vaso-occlusive crisis.
It is mostly seen for the first time when individuals are between 6 to 9 months old as this is the time when the fetal hemoglobin is being replaced with the mutated adult hemoglobin, causing the manifestations of the disease. It is one of the earliest presenting symptoms for sickle cell disease.
Symptom #4: Fever
Fever or pyrexia occurs when the temperature of the body is above the normal range due to the increase in the body’s set point of temperature. This increased set point causes a sensation of coldness and muscle contraction to produce and conserve heat.
In sickle cell disease, the presence of a fever is most likely due to an infection. Repeated episodes of vaso-occlusion in the spleen, can lead to splenic infarctions and autosplenectomy (shrunken and non-functional spleen). The spleen plays an important role in the body’s immunological response (i.e. an important source of antibody production). Thus, patients without a functioning spleen are susceptible to frequent severe infections caused by encapsulated pathogens (i.e. Streptococcus pneumoniae, Haemophilus influenzae). Also, patients with sickle cell anemia have an increased risk of Salmonella osteomyelitis (bone infection). This is why vaccinations are especially important to help prevent potentially life-threatening infections.
Symptom #5: Delayed Growth
Since sickle cell disease is a type of anemia, this means that the body has a decreased amount of red blood cells that function to carry and supply oxygen to parts of the body. The sickle-shaped cells cause the cells to become stiff and increases the likelihood of them being stuck in the blood vessels or destroyed by the spleen.
Since it is a genetic disorder that is hereditary, it is a chronic condition that is present since birth. Chronic and severe anemia can result in tiredness, pallor, delayed healing, and growth retardation. This can be seen in children with sickle cell anemia, who can be underweight, of short stature, and experience delayed sexual maturation (late puberty).
Symptom #6: Vision Issues
In patients with sickle cell disease, vision issues occur due to vascular occlusion, which may affect the retina, conjunctiva, iris, and choroid. One of the most significant and severe vision issues that affect sickle cell disease patients is proliferative sickle cell retinopathy (PSR). The risk for this condition increases with age, and the peak onset is between 15-24 years in men and between 20-39 years in women. Early stages of PSR are usually asymptomatic. It is recommended that patients with sickle cell disease have dilated funduscopic examinations (visualize the fundus of the eye) every 1-2 years by a retina specialist, beginning at age 10.
The treatment and management involved when vision issues occur is the prevention of vision loss from retinal detachment, vitreous hemorrhage, and reducing neovascularization in the lesion (formation of new blood vessels).
Symptom #7: Jaundice
Jaundice is defined as the yellowish pigmentation of the skin, mucous membranes and sclera of the eyes due to high levels of bilirubin in the blood. There are many causes of jaundice which range from mild to fatal conditions. Jaundice can occur when there is excessive red blood cell breakdown, consumption of certain foods, side effects of medications, gallstones, pancreatitis, leptospirosis, and more.
In sickle cell disease, the excessive breakdown of RBCs by the macrophages (immune system cells) of the reticuloendothelial system (spleen, liver, and lymph nodes) leads to high levels of unconjugated bilirubin. In short, protoporphyrin, an important component of hemoglobin in RBCs, needs to be broken down into unconjugated bilirubin and sent to the liver. Under normal circumstances, in order to be excreted in the bile, bilirubin must undergo a process of conjugation in the liver that makes it water-soluble. However, if there is excessive production of unconjugated bilirubin, the ability of the liver to take up and conjugate this product is overwhelmed. Thus, unconjugated bilirubin will accumulate in the bloodstream and cause jaundice.
Symptom #8: Chest pain
Patients with sickle cell disease may experience episodes of irreversible sickling of RBCs that can lead to complications of vaso-occlusion. One important site of vaso-occlusion during these episodes is pulmonary microcirculation, often precipitated by lower respiratory tract infections (pneumonia).
Adult patients with acute chest syndrome present with chest pain and shortness of breath. Most importantly, acute chest syndrome is a medical emergency because it is the most common cause of death in adult patients with sickle cell anemia.
Symptom #9: Pallor
Pallor refers to the paleness that can be seen in stressful situations, use of stimulants, emotional shock, illness, and anemia. Pallor is visible in the skin (i.e. palms and soles), mucous membranes, and the conjunctivae of the eye. In anemia, regardless of the cause, there is poor tissue oxygenation. Oxygenated blood is precious, thus its shunted away from the skin and other peripheral tissues, to permit enhanced blood blow to vital organs. This gives rise to pallor, an important physical finding in patients with anemia.
Besides anemia, other causes of pallor include vitamin D deficiency, lead poisoning, motion sickness, hypothyroidism, blood loss, and more.
Symptom #10: Dyspnea
Dyspnea refers to shortness of breath or difficulty breathing. It has been defined as a subjective experience where the affected individual experiences chest tightness, the increased effort needed to breathe, and the feeling of not having enough air. Anemia is a common cause of dyspnea, primarily due to a mismatch in oxygen supply capacity and demand. Furthermore, in severe anemia cardiac failure (due to a high output state) can also cause shortness of breath. In sickle cell disease, there is considerable overlap with other mechanisms than can also produce dyspnea. For example, as mentioned above, acute chest syndrome is a life-threatening complication of sickle cell disease, which can also cause shortness of breath.
Dyspnea occurs in cases such as cardiac ischemia, asthma, pneumonia, heart failure, anxiety, panic disorder, interstitial lung disease, and more.