What Is Phenylketonuria?


Phenylketonuria is a rare genetic disorder. An individual with this condition is unable to process an amino acid called phenylalanine. Amino acids are the building blocks of protein, and foods that are high in protein are also high in phenylalanine.
The prevalence of PKU varies across geographic regions and ethnicities around the world. People who are of European or Native American descent have higher rates of the condition than those from other ethnic groups. Most cases of the disorder are detected during newborn screening in the U.S. as well as a number of other countries.

1. What Are the Causes?
PKU is caused by a defective gene. This gene is partly responsible for breaking down the amino acid phenylalanine. This amino acid is present in numerous food sources and aspartame. When the body is unable to process the amino acid, it begins to build up.
As the phenylalanine accumulates, it causes numerous physical and mental health issues. When a baby is born with PKU, the primary source of nutrition is formula or breast milk, both of which are high-protein sources and contain phenylalanine.
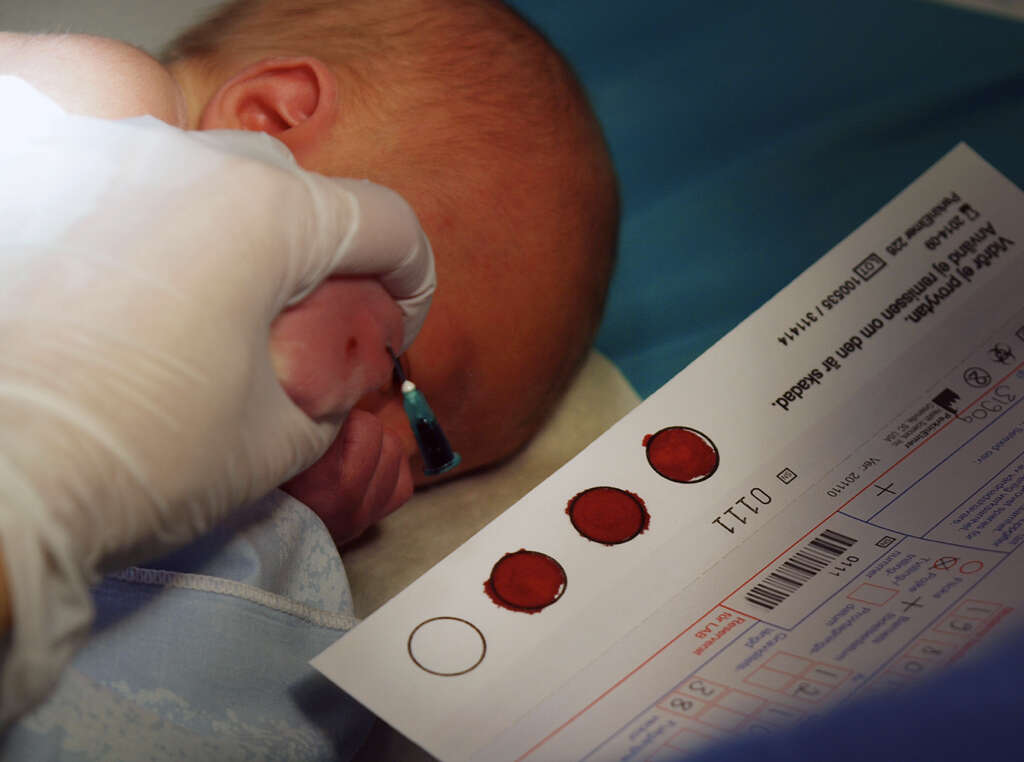
2. What Are the Inheritance Factors?
The disorder is an inherited condition that is present from birth. However, certain genetic conditions are required. PKU is an autosomal recessive disorder. As such, the baby must inherit the defective gene from both the mother and the father. A child who inherits the gene from only one parent becomes a carrier of PKU but does not have any of the symptoms.
An individual who has the disorder can pass it on to a child, but someone who is a carrier can also pass the gene on to offspring. Many times, people who are carriers do not realize it. The highest rates of PKU are found in those cases where both parents are unaware that they have the defective gene.

3. What Are the Symptoms?
Children who are born with the disorder display no immediate symptoms because they have not consumed protein-rich formula or breast milk. The symptoms only appear if treatment does not begin immediately or is not followed. They usually begin within the first few months.
Babies who develop symptoms frequently experience neurological issues such as seizures. They often have a musty smell to their urine, breath or skin. The growth of the head does not keep up with the growth of the body and mental development is delayed. Developmental delays continue as the child ages, and behavioral and emotional issues also arise.
4. How Severe Is PKU?
The severity of PKU varies from mild to severe. In the milder or more moderate cases, the body still produces some of the enzyme that breaks down phenylalanine; therefore, the degree of accumulation is lower. In these instances, the risk of developing severe symptoms declines. However, even in these less severe forms, the best outcomes require treatment.
Classic PKU describes the more severe form of the disorder. This classic form results in little to no enzyme production, significantly increasing the risks of severe symptoms when treatment is not followed. If treatment is maintained, then the likelihood of developing symptoms is low.

5. What Are the Potential Complications?
The importance of following prescribed treatment does not end after childhood. Older children, teens and adults who do not adhere to diet and other recommendations experience complications from too much phenylalanine in the system. Intellectual and developmental disabilities that can develop beginning in the first months are irreversible.
There is the potential for neurological issues ranging from tremors to seizures. Older children and adults can still develop significant social, emotional and behavioral problems, as well as other health issues. Adult women who have too much of the amino acid in their blood during pregnancy place themselves and their babies at risk.

6. What Are the Effects During Pregnancy?
When a woman with PKU is pregnant, the potential for maternal PKU exists. This condition results in exceptionally high levels of phenylalanine in the blood, even in those who have milder forms of the disorder. When the PKU diet is not followed prior to and during pregnancy, the chances for miscarriage and birth defects are increased.
Those children who are born to mothers who have elevated phenylalanine do not frequently inherit PKU. However, there is an increased risk of birth defects. Microcephaly (an abnormally small head), low birth weight, facial malformity, developmental delays, heart defects, intellectual disabilities and behavioral issues are all possible outcomes for the baby.

7. When Should You See a Doctor?
Newborn children in the U.S. are automatically screened for PKU. If your child is diagnosed with the disorder, your doctor will discuss a treatment plan with you. Beginning the PKU diet immediately is important in preventing the long-term damages the disorder causes.
If you are a woman who has PKU and plans to have children, following the special diet prior to becoming pregnant and during pregnancy is imperative to the wellbeing of your child. Contact your health care provider for guidance. Adults with the disorder should maintain regularly scheduled doctor’s visits and follow the diet over the course of their lives.

8. How Is PKU Diagnosed?
If you have a family history of PKU, your doctor can screen you before you have children to determine if you are a carrier. The screening is accomplished through a blood test. Doctors often also recommend that women with PKU and carriers be tested during pregnancy as well.
For newborn babies, the standard screening involves collecting a small blood sample shortly after birth, generally within the first two days. The test for the disorder is conducted simultaneously with tests for other metabolic disorders.
9. What Is the PKU Diet?
The primary form of treatment for PKU is dietary. For those with the condition, it is necessary to severely limit high-protein foods such as meat, milk, cheese, eggs, nuts, beans and legumes. It may also be necessary to limit consumption of vegetables that contain higher levels of protein, such as potatoes.
Doctors often recommend consuming just enough protein to support normal growth and health. However, consumption of a specialized formula is often necessary for infants, children and adults, especially those with classic PKU. The dietary needs of any one person depend on multiple factors and can vary over a lifetime, making ongoing care and evaluation necessary.

10. What Other Treatment Options Are There?
Adults with PKU may have an additional treatment option. There is a supplement called neutral amino acid therapy, available in both powder and tablet forms, that may reduce the absorption of phenylalanine into the blood. This option is not necessarily the right choice for everyone, but those who are interested should discuss the possibility with their physician.
A relatively new drug, saproterin, has been approved by the FDA. This medication has the potential to increase phenylalanine tolerance for some people. Future scientific studies are needed to determine long-term efficacy and safety.








